Fast, Flexible eCOA for Global Trials
Streamline Patient Data Capture. Deliver High-Quality eCOA.
Clinical trials are more complex—and more critical—than ever. YPrime’s electronic Clinical Outcome Assessment (eCOA) platform is purpose-built to help you move faster, capture better data, and meet patient needs globally. With advanced flexibility, scalability, and intuitive design, YPrime gives your team the tools to deliver confident results at every stage of the trial.

")
Configurable, Scalable eCOA That Adapts to Your Protocol.
Every study is different. YPrime’s flexible eCOA platform adapts to your protocol, region, and patient population—without slowing you down.
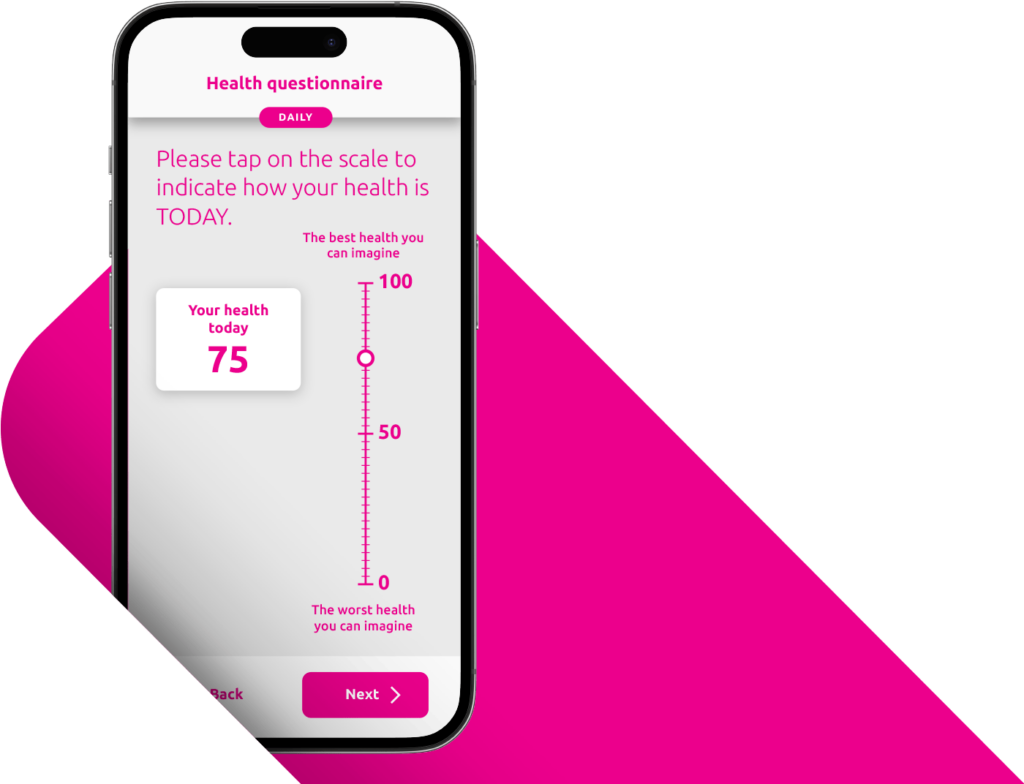
A Patient-Centered Approach Drives Compliance and Data Quality.
A positive patient and site experience leads to stronger compliance and better data. YPrime’s patient app is developed with real-world usability research to support participants everywhere.
eCOA Reporting That Powers Decisions.
Turn raw clinical data into near real-time insights that drive smarter oversight, faster interventions, and better outcomes. Traditional eCOA platforms leave sponsors waiting for answers. Built with global inspection-readiness in mind, YPrime’s built-in reporting tools give you instant access to the information you need to act decisively throughout your trial.
Want a preview? Book a demo of our eCOA reporting dashboards.
Future-Ready eCOA—Built for What’s Next in Clinical Trials.
47% faster study startup
100+ countries supported
BYOD and provisioned device ready

YPrime has a clean and seamless UI. It is able to take the data and put it into graphs and figures to show where the data is trending.
Experience That Matters.

~1,000
Studies Implemented Globally

19+
Therapeutic
Areas

14+
Years eClinical Technology and Services
Future-Proof Your Trials with
Adaptive, Innovative eCOA Technology.
Clinical research is evolving. With YPrime, you’ll be set up for success with eCOA built to adapt to industry changes. Let’s get started!
Explore Insights from Our Experts.
Gain valuable perspectives on clinical trial design, high-quality data capture, operational efficiencies, and, ultimately, how to solve with certainty in clinical research.


